���� �˹����ܼ�������ǰ��δ�е��ٶ���ҽ����е����Ӱ����ϡ�����Ԥ�⣬�����廯������ջ����ƣ�AI�ѳ�Ϊ�ƶ�ȫ��ҽ�ƿƼ����ĺ��Ķ����������㷨�Ӹ�����������ΪӰ���ٴ����ߵĹؼ�����������ϵ���ݻ�Ҳ��Ӧ������ˮ����������ڴٽ�������ȷ����ȫ֮���ҵ�ƽ�⣬��Ϊ������ܿ�ܵĺ������⡣ ��ȫ��Χ�ڣ�������ܻ�����½�������̽��AI/MLҽ����е�ļ��·������ŷ����������صġ�˫��ʽ���ģʽ������ǰ�С�ŷ�˲�δ���˹�����ҽ����е��Medical Device Artificial Intelligence, ��MDAI������Ϊ��һ��ܶ����Dz��ú��ݵ��ӵ���ϵʽ������ʽ��ҽ����е��ȫ������Ҫ����MDR/IVDR�������㷨�������������Ȩ��������AIA���ܣ�������ϵ��ŷ����������ܣ�NLF����ԭ����Эͬ�������γ�ȫ����ϵͳ����MDAI��ܿ�ܡ� �ڴ˱����£�����ŷ��MDAI�����ϵ�ı߽硢����Ϲ�Ҫ���ѳ�Ϊ����ϣ������ŷ���г��Ĵ�����ҽ����ҵ��ǰ�ᡣ����ּ���Է���֮������ù�ϵΪ����㣬��MDR/IVDR�Ļ�������AIA�ĺ�����ơ�MDAI�Ľ綨������϶����ƣ��������ļ�����������������ල�����к���Ⱦ���Ϲ�Ҫ��ϵͳ����MDAI��ŷ�˵������Ϲ�·��������������й���ҵ��ʵ�����顣 1. MDR��IVDR�ļ�ܺ�����
��ŷ�˵ļ����ϵ�У�ҽ����е��Medical Devices, ��MD�������������ҽ����е��In Vitro Diagnostic Medical Devices, ��IVD�����ֱ��ܵ��������з����Լ������ҽ����е���桷��The regulation on medical devices, Regulation (EU) 2017/745, ��MDR�����롶�������ҽ����е���桷��The Regulation on In Vitro Diagnostic Medical Devices, Regulation (EU) 2017/746, ��IVDR���������������湲ͬȡ�������ڵ�ָ����ϵ��MDD1��IVDD2����������ŷ��ҽ����е��ܵ���������������˹�����������ҽ�Ʋ�Ʒ���ԣ�����MDR��IVDR�Ļ��������ǰ��պ���ŷ�ˡ��˹����ܷ�������Artificial Intelligence Act, ��AIA������������ǰ�ᡣ
1.1 ҽ����е��������ϵļ�ܱ߽�
MD��IVD�ķ�Ұ������Դ����е��;�IJ�ͬ��
�� MD��ָ���������ڲ���ֱ��������������ҽ����е��������ϡ����ơ���⡢����������������ܵ��豸�������������
�� IVD��ָ����������������������ѪҺ����Һ����֯������е���Լ�����������Ŀ�����ṩҽѧ��Ϣ��֧����ϻ�Ԥ�⡣
MDR��IVDR���ߵ������������ƣ���ǿ������ȫ�ԡ����ܡ������ԡ�����֧������������IJ�ͬ��MDR��ע������ֱ�Ӱ�ȫ���ٴ���Ч����IVDR���ǿ����ѧ��Ч�ԡ������������ٴ����ܵ���ʽ��֤��
��AIҽ�Ʋ�Ʒ���ᄈ�У���һ�������ۻ��ġ����磬һ������AI�㷨���ĵ��ź������������MDR��ܣ���һ��AI�㷨���ڻ�������������ϵͳ��������IVDR��Ͻ��Χ����ˣ��ڽ���AI��Ʒ�ġ��ʸ��жϡ���qualification��ʱ������Ӧȷ����������;�������������屾����������������������������һ������˲�ͬ���������ѡ��·����
1.2 ���շ�������빫�������NB����ɫ
MDR��IVDR�����û��ڷ��յļ��ģʽ������Ʒ���յȼ�Խ�ߣ��Ϲ�Ҫ��Խ�ϣ�
�� MDR����е��Ϊ�ļ���I��IIa��IIb��III����¼ VIII������������ۺϿ�����е�������ԡ���;������ʱ�估�Ի��ߵ�DZ��Ӱ�졣
�� IVDR�����ݸ�¼VIII����Ʒ����ΪA��B��C��D�ļ�������ά�ȸ���������������������Ϲؼ��ԡ����磬�漰��Σ��Ⱦ�������Լ�����HIV���Ҹμ�⣩���� Class D��������־����Ŵ����ȶ���AI��������㷨�������Class C��
�ںϸ�����·���ϣ������·�����������������������Notified Body, ��NB������Ϊ�����ļ�����鷽��������ԣ�NB���������ܻ�����һ���֣����Ƕ�������֯����ŷ��������Ա�������ܻ���ָ����������ŷ�˷���Ҫ��������ϸ�����������£��������CE��־�IJ�Ʒ���з���������������MD��IVD���ԣ�NB�ķ��ɶ���Ϊ�����ݱ�����ָ���ĺϸ�����������(a conformity assessment body designated in accordance with this Regulation)3��
�ھ�ָ��ʱ����MDD/IVDD������IVD�������������������ϸ����·����£���һ������֤ʱ���Ѿ��ս�4��
�� ����MDR�����и߷�����е��IIa�����ϣ��辭NB��飻
�� ����IVDR������ͷ��յ�Class A������Ʒ�⣬��������IVD����ҪNB���롣
������ϵ�仯ֱ�ӵ�����ŷ��NB��Դ�Ľ��ţ�Ҳ�ƶ�����ҵ����������NB��ͨ������MDAI���ԣ�NB������ע��ͳ��ȫ�����ܣ���������㷨ѵ�����ݡ�ģ����֤�������������ڹ������ɽ����Ե����ݡ�
1.3 MDR��IVDR�ĶԱ�
MDR��IVDR���������������ķ�����ϵ�������趨����ֵ���嵥�������ڲ�ͬ���͵IJ�Ʒ�����������ߵ���Ҫ����

�����ͼƬ�ɷŴ�鿴��
2.
���ҽ����е���ж���Ӧ�ó���
2.1 ������Ϊҽ����е��SaMD/MDSW��
��������������������е��ձ�ʹ�ã�ŷ�˼����ϵ�ԡ�������Ϊҽ����е����Software as a Medical Device, ��SaMD������ҽ����е��������Medical Device Software, ��MDSW���������˶���������ϵ��
MDR��2����ȷ�涨����������Ԥ����;���ڡ�����������ͼ�����ṩ��ϻ����ƾ�����Ϣ��������Ϊҽ����е��ŷ��ҽ����еЭ��С�飨Medical Device Coordination Group����MDCG�������䷢����ָ��MDCG 2019-115�У���һ��ϸ�����жϱ�����ָ��Ҫ�������̽�����������������жϣ�
(1) ȷ�ϲ�Ʒ�Ƿ����ָ�϶ԡ��������Ķ��壬���䲻�����������룬����������ָ�ϡ�
(2) �����Ƿ���ΪMDR��¼XVI��Ʒ��ҽ����е����ɲ��ֻ������Ƿ�������Ӱ��ҽ����е��ʹ�ã�����Ϊ�ǣ���ֱ�ӱ�����MDR/IVDR��ܡ�
(3) ���������Ƿ�ִ�г��������洢���鵵��ͨ�š�����������ѹ���IJ����������Ƿ��ҽѧ��Ϣ���з������������ͣ���ִ�д������������ܹ���MDSW��
(4) �ж�����������Ƿ�ּ�ڷ����廼�ߡ����ٴ����ߣ�����������ϡ�������������ľ��ߴ�����Ϣ����������Ⱥ�����ݻ��ܡ����л����в�ѧ��;��������ҽ����е������
(5) ��ǰ����������ۺ��жϣ���ȷ�ϸ������Ƿ�����ָ�϶�ҽ����е�����Ķ��壬�Ӷ��������Ƿ�Ӧ��ѭMDR/IVDR�µķ��շ�����CE�ϸ���������
��ˣ������Ƿ�����ҽ����е������������������Ƿ���ҽ��Ŀ�ķ����ڸ��廼�ߵ��ٴ���������AI���������ڽ�ѧ�о���������������Ϣ������ͨ�Ź��ܣ���PACSϵͳ��ҽԺ�Ű�������������ҽ����е��������������ҽ��������ϡ�����Ԥ������ƾ��ߣ��������MDR��IVDR�ĵ�����
���磺
�� �������ѧϰģ�ͷ��� CT Ӱ��ʶ��ν�ڵ�ϵͳ��������MDR��
�� �����㷨��ѪҺ��������ж������͵�ϵͳ��������IVDR��
�� ����ҽԺ������������ͳ�Ʒ�����AI���ߡ���ͨ��������ҽ����е��
ֵ��ע����ǣ�ŷ�˷���ǿ������ͼ��;��intended purpose�����ı���Ȩ�������̣�����ܻ��������˵���顢�û����漰�㷨������ݣ����ж�����ʵ���ܡ�
2.2 �˹�����ҽ����е��MDAI��
�˹�����ҽ����е��Medical Device Artificial Intelligence, ��MDAI������ŷ�˷�����ϵ�в���һ�������ķ��ɷ��࣬����ŷ���˹�����ίԱ�ᣨArtificial Intelligence Board, ��AIB������MDCGΪ˵��AIA��ҽ����е�������ù�ϵ������Ĺ����Ը��
��ǰ����������MDR��IVDRͨ���ԡ�������Ϊҽ����е��MDSW/SaMD�������ʸ�����չ���Ҫ���Ѿ�һ���̶��ϸ�������������صķ��գ�����ȱ����AIϵͳ���з��յĹ���6��Ϊ�ֲ���һ�հף�AIB��MDCG�����ָ���У���AIA���롰AI system���Ķ��壬�����MDR/IVDR���綨��ҽ����е��Χ������һ���漯���е��˹�����ϵͳͳ��Ϊ�˹�����ҽ����е��MDAI����
���ϣ�MDAI��ָ��MDR/IVDR��ܿ����ʹ���ҷ���AIA�������AI system���˹�����ϵͳ������ϵͳ��������MDR/IVDR�����ٴ�����ȫ�����ܷ����Ҫ��Ҳ����ѭAIA �Ը߷���AIϵͳ������������������ȡ�����ල����չ����ȸ�������MDAI�ĸ���綨ּ��ͳһ������һ���˹�������Ϊҽ����еʹ�á��Ľ���������ȷ��ŷ�˼����ϵ����˫�غϹ����ԡ�
3.
AIA�ĺ����ܣ��Ը߷���MDAI��Ҫ��
��MDR��IVDR��������������ϵ֮�ϣ�ŷ����2024����ʽͨ��ŷ�ˡ��˹����ܷ�������Artificial Intelligence Act, ��AIA������ȷ����ȫ����ȫ����˹�����������ܡ��ⲿ����ij�̨����־��MDAI�ļ�ܽ��롰�ݺ���ӡ����½Ρ�����ҵ����Ҫ����ҽ����е����İ�ȫ������Ҫ��Ҫ����AIA�Ը߷���AIϵͳ�������������������
3.1 AIA�Ķ�λ����MDR/IVDR��Э������
AIA��Ϊһ�������������������������ŷ���г����ṩ��ʹ�õ�AIϵͳ���������ṩ���Ƿ�λ��ŷ�˾��ڣ������Ŀ������ȷ��AIϵͳ�İ�ȫ�ԡ�������ɽ����ԣ���ֹ�㷨������������ƫ�У�AIA���ƶ���־��ŷ�����˹�������������������ϵͳ������յ�����½Ρ�
��ǰ����������MDR��IVDR��Ҫ���Ѷ�SaMD/MDSW�ķ����������꾡�涨�������߲�δ���MDAI�����е��㷨���ա���̬ѧϰ����������ƫ�е����⽨����ȷ�ļ�ܿ�ܡ�AIA�������ֲ�����һ��ܿհף���ͨ��ȷ����AIϵͳ�ڽ�������ȫ������Ȩ������DZ��Σ����ʶ������غϹ�Ҫ�Ӷ���MDR/IVDR���в��䡣
����ŷ����������ܣ�New Legislative Framework, ��NLF����ԭ��7�����ڰ���һ�������߷����˹�����ϵͳ��ҽ����е��Ʒ��MDR/IVDR��AIAӦ���������á�����䡣
�Դˣ�AIA�ڵ�8����2����ڡ��Ը߷���AIϵͳ�ĺϹ�Ҫ����������ȷ��
�������Ʒ����һ���˹�����ϵͳ������������Ҫ���Լ�����һA������ŷ��ͳһ������Ҫ�������ڸ�ϵͳ�����ṩ��Ӧ����ȷ�����Ʒ��ȫ�������õ�ŷ��ͳһ��������������Ҫ����ȷ����1�����ἰ�ĸ߷����˹�����ϵͳ���ϱ��ڹ涨��Ҫ��ʱ��Ϊ��ȷ��һ���ԡ������ظ�����С�����⸺�����ṩ�߿�ѡ�����齫���ṩ���й����Ʒ�ı�Ҫ���Ժͱ������̡���Ϣ���ļ����ϵ�����һA������ŷ��ͳһ����Ҫ��������ļ��ͳ����С���
ŷ����ؼ��ָ���ļ�Ҳָ����
��Ϊȷ��һ���ԡ������ظ�������ȵؼ��ٶ��⸺����MDAI�����̣�����AIA��8����2��Ĺ涨����Ȩѡ�����ʵ�����½������MDAI���ṩ�ı�Ҫ�����뱨�����̡���Ϣ���ļ������������Ѹ���MDR/IVDR�������ļ������֮�С�ǿ�ҹ���MDAI����������AIA��8����2�����ṩ������ԡ�Ȼ���������ø��������ʱ��MDAI������Ӧȷ����MDAI��ȫ����AIA��MDR��IVDR���κ���������֮ŷ����������������Ҫ��
3.2 AIA��6�����߷���AIϵͳ�����ʽ�ж�����
AIA��6����ȷ��һ��AIϵͳ�Ƿ����ڸ߷���AIϵͳ��High-Risk AI System�����жϹ������IJ�δ���о���ҵ���Ʒ������ͨ��һ�����ʽ�ж����ƣ�����AIϵͳ�ļ������������ŷ�˲�Ʒ��ȫ��������MDR/IVDR�����νӣ��Ӷ�ʵ�ַ��շּ����Զ�ʶ��
���������������£�

�����ͼƬ�ɷŴ�鿴��
���嵽ҽ����е����AIA�ĸ���һ�涨�ˡ�ŷ��ͳһ�����嵥��������A�ڵ�11�κ͵�12����ȷ������MDR��Regulation (EU) 2017/745����IVDR��Regulation (EU) 2017/746������ˣ����ǡ���Ϊ��Ƕ��ҽ����е���������ҽ����е�������е�AIϵͳ��һ�������AIA��6����һ����Ҫ��ġ��������ԡ����������ڵڶ����ġ�������ԡ������ò�Ʒ�Ƿ���Ҫ����NB���е������ϸ���������ȡ��������MDR��IVDR�µķ��յȼ����ࡣ��ͬ���յȼ���Ӧ��ͬ�����Ҫ��Ҳ�ɴ˾�����AI�Ƿ������϶�Ϊ���߷���AIϵͳ����

�����ͼƬ�ɷŴ�鿴��
��ˣ���MDR��IVDR����£����������и߷��յȼ���ҽ����е���䱸��Ƕ���AIϵͳ�����Զ������߷��ջ��ƣ����������������������ϸ�ĵͷ��ղ�Ʒ�����ܻ��⡣����֮����ҽ����е�����������еһ����ҪNB����ϸ�����������Ƕ���AIģ�鼴�Զ�����ΪAIA�����µġ��߷���AIϵͳ������ͬʱ����MDR/IVDR��AIA��˫�ؼ�ܡ�
���������̶��ԣ�����ζ�ţ������ٵ����ж�MDAI�Ƿ߷��ա���������������������ϵ�ͼ����ĵ���ϵͳ������AIA�Ը߷���AIϵͳ��Ҫ��ĺϹ�����
4.
�߷���MDAI�ĺ�������
��AIA��8��15��Ϊ�����ߣ����߷���MDAI��Ҫ������Ƕ��MDR/IVDR��ܼܹ�֮�У���Ҫ�γ��������ĸ�����ĺϹ�ӳ�䣺
4.1 ������ϵ��Management Systems��
�߷���MDAI�ļ����ϵ��MDR/IVDR��AIA���γɻ�����֣���ͬȷ��ϵͳȫ�������ڵİ�ȫ�ԡ�����һ������Ի���Ȩ���ı�����
���ȣ����������ڹ������棬MDR��IVDRҪ����������������Ʒ���������ڽ��������ļ����������ƣ�AIA��һ��ǿ������һҪ����ȷ�߷���AIϵͳ������ơ����������ԡ������µĸ��ν���ϵͳ�������ල�������ɳ������е��������ڹ�����ϵ���ر�����Ծ߱�����ѧϰ������MDAI��AIA�涨Ӧͨ�����к�����ϵʵ�ֶ�̬����ʶ��������һ����ά������ȷ����Ҫ��Ϣ�����������ߡ�������ܻ�����
��Σ�������������ϵ��Quality Management System, ��QMS�������棬���������Ҫ�������̽���ϵͳ�����ļ����������Ľ�������������ϵ����ȷ�����Ϸ����뼼��Ҫ���ڴ�ͳҽ����е���ʵ���У�MDR/IVDR��δ��QMS�ļ�����ϸ��������ȷ�涨����ͨ��ͨ������Э��������ʵ�ֺϹ���أ�����ISO 1348511��Ϊҽ����еQMS����ͨ�б����ѳ�ΪNB�ڿ�չ����������ʱ�ձ���õĻ�����ܡ�AIA��17����ȷ�ṩ���뽨�����Ƿ��չ������������������к��⼰�¼������Ҫ�ص�QMS������������MDR/IVDR�µ���ϵ�������ϣ��Լ����ظ�������һ���ԡ����⣬��Ը߷���MDAI������������ϵ��ŷ������Э�������ƶ�����Ҳ����ͬ�������С�
��������չ�����ϵ���棬AIA��MDR��IVDR��Ҫ�������̽�����ʶ���������ķ��գ���������չ�����ϵ�д����ɺ�������ķ��ա�AIAǿ��ͨ��ȫ��ķ������������ջ����ʩ���ļ���¼�Լ���ϵͳ���ܵij�����⣬ȷ���߷���MDAI��ҽ�ƻ����еİ�ȫ����ɿ��ԡ��߷���MDAI�������̿ɽ�AIA��9�������MDAI�涨�ĸ��ӷ��չ���Ҫ����ͬAIA�涨�IJ��ԡ��������̼������Ϣ���ļ���������������MDR��IVDR�ѽ����ķ��չ��������֮�С�
4.2 ����������Data Governance��
AIA��MDR/IVDR����MDAI���������������˹��ƣ��������÷�Χ���ص�������ͬ��MDR��IVDR������ҽ����е�ٴ����ݵĿ�ѧ������Ч�ԣ���AIA���ע�˹�����ϵͳ�����ݵ���������������ƫ����أ���ȷ���㷨���еİ�ȫ��Ϲ档
���ȣ�MDR��IVDRȷ���������ٴ�֤�ݵ���������Ҫ��������MDR/IVDR�Ĺ涨��������е�������ٴ����ݱ����Ƚ���robust�����ɿ���reliable����������Դ��������õ��о���derived from well-designed studies����Ϊ֧�ֳ���ٴ�֤�ݵ����ɣ��ٴ�������clinical evaluation, ������MD��������������performance evaluation, ������IVD�������������еԤ����;������ٴ����ݡ�������֤��ȷ�ϼ�������е��Ƶij����뼼�����Լ��ɴ˲������ļ�����Ϣ����Ӧ��Ϊ�ٴ�֤�ݵ�һ�������Լ�¼������NB������顣
��Σ�AIA��10����ǰ�������Ͻ�һ����������Ը߷���AIϵͳ��������������������涨������ѵ������֤�����MDAI�����ݼ�Ӧ�߱�����ԡ���ִ����ԣ����������ܷ�Χ�����������Ҿ����ʵ�ͳ���������������轨��ϵͳ�������ݹ������������ݻ�ȡ����������ע���洢�������������õȻ��ڣ�ȷ���������������������ڵ������������ԡ�AIAͬʱҪ��������ʶ��ͼ������Ӱ�콡������ȫ�����Ȩ����ƫ���ֹ���㷨���������������IJ�ƽ�Ƚ����
���⣬AIA��ͨ����¼��������־��¼����ǿ�����������Ŀ����ԡ��߷���MDAIӦ�߱������������������Զ���¼�¼���logs������������ʶ��������ƫ���ϵͳ���µ��µ�DZ�ڷ��գ�����Ϊ���к�����ϵ����Ҫ��ɲ��֡���Ҫ����MDR/IVDR���ٴ����ݳ��������������νӣ��γɱջ���ܡ�
������漰��������ʱ��AIAҪ�������̵�����������ʩӦ��ȫ���ϡ�ͨ�����ݱ�����������GDPR�������������ռ�Ŀ�ġ�������ʽ���Ϸ��Ա������������̿�ͨ����������֤������֤���������ĺϹ��ԣ��Ӷ�����������������Ч�ʡ�
���ϣ�AIA�����㷨���������������ƫ����ƣ�MDR��IVDR��۽��ٴ���������ݿɿ�����������֤����ͬȷ���˸߷���MDAI������������������������������������ݵ�ʹ�á�ϵͳ��������������ƫ����ػ��ƣ��Լ�ȫ�������ڵ����ݿ��������Ϲ�Ҫ��
4.3 ����������ල��Transparency & Human Oversight��
4.3.1 ��������
��ŷ�˼�ܿ���У�AIA��MDR/IVDR����������Ϊ�߷���MDAI�Ļ���������ͨ�������Ļ�����ϵͳ��ơ�������ʹ�ø���ʵ����Ϣ�ɻ������������ԡ�
���ȣ�AIAͨ����������ȷ�����ȵ��ƶȽṹ��
һ���棬AIA��13��12Ҫ���ṩ��ȷ��ϵͳ���о��С��㹻���ȡ������ṩ����������ʹ��˵�����Ա㲿��������֪����ߡ������˵��ϵͳ��Ԥ����;����ֹ��;�����ܱ߽��Լ��˹���������Խ������ɹ��̵Ĺ��ס����⣬�����漰ֱ������Ȼ�˽�����ϵͳ�����£�AIA��50��13�ر�Ҫ���û���ȷ����֪������AI���������Ǹ���ʵ�Ժ����û������ѿ�����ʶ��
��һ���棬AIA��10��14�涨�˸߷���AIϵͳ��������������Ҫ��ѵ������֤��������ݾ߱�����ԡ������ԡ������Բ��������ܷ�Χ����ƫ�������Ҫ�������̽������ݹ������ļ������̣�ʹ��ܻ����Ͳ������ܹ�����AI���ܵĻ������ݽṹ����һ���أ�AIA��12��15Ҫ��߷���ϵͳ���ֳ�ֵ���־��¼����֧�֡����ܿ����ԡ���functional traceability�����������к�ල��������ʵ��
��Σ�MDR��IVDRͨ������I��ͨ�ð�ȫ������Ҫ����General Safety and Performance Requirements, ��GSPRs����16ǿ����������
GSPRһ����Ҫ���������ṩ������еԤ����;�����з�ʽ�����ռ����Ƶij����Ϣ��������������AI���������е���ܵĹ���ʽ����һ������Ҫ�����������ѭ��ǰ����ˮƽ���������������ڹ��������չ������̣����ƶ���ȷ���ɽ��������ļ������ԡ����⣬����MDR/IVDR����II����III�����������ڼ����ļ�����ϸ˵��AI������������з�ʽ���������ݴ�����ʽ��������ɻ��ƣ�ʹ�����ߺ�NB�ܹ�����AI����е����������Ӱ�졣
ͨ���������ƣ�MDR/IVDR��AIAЭͬȷ��NB�����������û��ܹ�����߷���MDAI�����ʡ����з�ʽ��������ԣ�֧��֪��ʹ��������顣
4.3.2 ����ල����
������ල���棬AIA��MDR/IVDR��ͬȷ�����ԡ��˹����տ��ơ�Ϊ���ĵİ�ȫ���Ͽ�ܣ�Ҫ��߷���MDAI����ơ�������ʹ��ȫ�����б��ֿɿ��ԣ�ʹ�˹�������Աʼ���ܹ����⡢��Ⲣ�ڱ�Ҫʱ��Ԥ��ϵͳ���ߡ�
����AIA��14����1��17���߷���MDAI������뿪�����������ʵ�������ල���ƣ�ʹ�˹�������Ա�ܹ����⡢��Ⲣ�ڱ�Ҫʱ��Ԥϵͳ��Ϊ����2��18��һ��ȷ�ϣ�����ල��ɷ��չ����еĻ����ʩ��AIA�����Բ���19ҲҪ��������Ӧȷ��ϵͳԤ��IJ���Լ�����ñ�ϵͳ�������ǣ���ϵͳ�����ܹ����˹�����ָ��������Ӧ������ζ�Ÿ߷���MDAI�ڹؼ����߹����б���ʼ�ձ����������Ԥ�Ŀ����ԣ�ȷ���˹��ж���Ϊ���տ�����Դ��
��MDR/IVDR����£���Ҫͨ��Ҫ��MDAI�����Ӧ�ﵽһ����GSPR��֧������ලԭ��Ϊȷ���߷���MDAI�İ�ȫ����Чʹ�ü����𣬲�ʹҽ����Ա��ҽ�ƻ����ܹ�ʵʩ�ʵ��ල������Ҫ�ල���ơ��������Ƽ���Ԥ��ʽ��ʹ��˵������ѵ���ϼ������ļ�����ȷ�ļ�����ʹ�˹��ල�߱��ɲ����Բ�������е�İ�ȫ�����ܱ�����ϵ��
4.4 ȷ�ԡ��Ƚ��������簲ȫ��Accuracy, Robustness and Cybersecurity��
MDR��IVDR��AIA���������ȷ����ͳһ�ļ��ԭ���߷���MDAI������ǰ�����к�Σ�����߱��Ƚ������簲ȫ��ʩ����ȷ�����ձ��Ͽɵļ���ˮƽ��ʵ�ֶԹ��ڽ����Ͱ�ȫ�ĸ�ˮƽ��������ܿ����ȷָ�����κ�����е������صķ��ն���������ڿɽ��ܷ�Χ�ڣ���Ҫ������������е���п��ܵ�����ģʽ�£�����Ч�������֮����ʵ�ƽ�⡣Ϊ�ˣ������̱��볬Խ��һ�Ĺ��ܰ�ȫ�ӽǣ��ۺϿ�������е��ȫ����safety���롰���簲ȫ��������security��֮���������ϵ��ȷ�������ڷ��տ�����ʵ��Эͬ20��
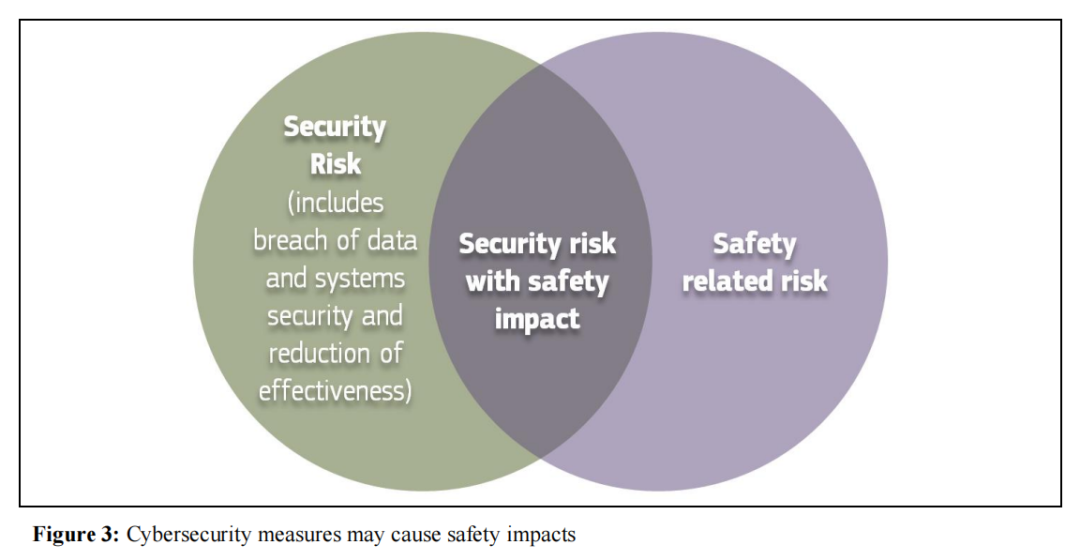
21
�����ͼƬ�ɷŴ�鿴��
���MDR��IVDR��AIA�ľ���涨���������������ʵ�֡������ʲ�������������ϵ�ں�����ά�����д�������
���ȣ��������������棬������ʵʩ�����簲ȫ��ʩӦ��������ԣ�ּ�ڷ�ֹδ����Ȩ�ķ��ʡ����繥����ϵͳ���������ݴ۸ģ���ȷ��ϵͳ���������ԡ��ر���AIAҪ��߷���MDAI�����ȡ�ض��ļ����ֶΣ���Ӧ���˹��������еĴ�����22�����ֱ�����������ϵͳ�����������������˹������ض��ʲ�����ѵ�����ݼ�����ѵ��ģ�ͣ��Լ��ײ����ͨ������ICT��������ʩ23����Σ��������������棬������Ӧ��ȡ��ʩ�������ݴ�����洢�İ�ȫ����ֹ������ģ��Ͷ�������߱������Ӧ�����簲ȫ�¼�����������ЩҪ��ᴩ��MDAI������ȫ���̣�������ѵ������֤�Ͳ��Խ���ʵ��Ӧ�İ�ȫ������ʩ������ڹ�����ϵ���棬�������簲ȫ�Ǹ߷����˹�����ϵͳ�Ļ���Ҫ�������̵ķ���������ش�ʩ����������չ�����ϵ������������ϵ���������迪չ����������ʶ��©����ʵʩ�����ʩ24����������ϵ������ܺϸ�������������25��
5.
�Ϲ�·�����й���ҵ��Ӧ��
5.1 MDAI�Ϲ��ͼ���ӿ��������к��ȫ���̹���
��ǰ�ĶԸ߷���MDAI��������ĺϹ����������ϵͳ�������Կ�������ЩҪ��ᴩ��Ʒ����ƿ�����ʵ��ʹ�õ�ȫ�������ڡ�Ϊ����������ЩҪ���ڿ���������ǰ�����к�εľ���ֲ����νӣ����½��ԡ��Ϲ��ͼ������ʽ�ܽ�MDAI��ȫ���̼��Ҫ�ؼ��������Ϲ��ܣ�


�����ͼƬ�ɷŴ�鿴��
5.2 ������Ӫ������ŷ��ɫ����
����MDR/IVDR��AIA��ͳһ��ܣ���ŷ��Ͷ��MDAI��Ʒ���й���ҵ���뽨�������ľ�����Ӫ����ϵ�����������̣�Manufacturer����ŷ����Ȩ������Authorised Representative, ��AR�����������̣�Importer���ͷ����̣�Distributor�����Լ��Ϲ渺���ˣ�Person responsible for regulatory compliance, ��PRRC������
�������Ƿ����ϳе�ȫ���Ϲ���������壬��ȷ����Ʒ����MDR/IVDR��AIA��Ҫ��İ�ȫ�ԡ����ܡ��������������ȡ�����ල���������簲ȫ����Ҫ��������ͬʱ��AIA�����µĸ߷���AIϵͳ�ṩ�ߣ�Provider���������м����ļ�����־��¼�����к��⼰�ش������������Ρ�
ŷ����Ȩ������AR���Ǿ�����ҵ���õĹؼ���ɫ��������ŷ�˾��ڳе��������Σ������ܻ��ع�ͨ����ȷ�������ļ����ϸ�������CE��־����Ҫ��AIA��δǿ������AR����NB�������ͨ��Ҫ������ҵͨ��AR���о������Ρ�
��������������������Ʒ�Ƿ��עCE��־���Ƿ����EUDAMEDע�ᡢ��ǩ��UDI�Ƿ����MDR/IVDRҪ���Լ��ڹ�Ӧ���мල��Ʒ��ȫ�ԡ��κζԲ�Ʒ�Ϲ��Ե����ʶ����ϱ����ܻ��ز���������Э��������
���⣬����MDR/IVDR��15���������̻��������Ϲ渺������PRRC�����ල�ϸ����������к���ͼ����ļ��ij���ά����
������ɫ�����й���ҵ����ŷ���г���������á�
5.3 ����ս�Խ���
MDAI��ŷ�˺Ϲ治������������꣬����ҵ�������ϵ�����������ĸ���Ҫ��
���ȣ�AIAҪ�������̽�������ģ�Ϳ�����ѵ������֤�����ԡ�������������־��¼��������ƫ�п��Ƶ���ǿ������������ϵ��QMS�����й���ҵӦ������ISO 13485�Ļ���������AIAҪ���γ�ͳһ��AI�Ϲ���ϵ��
��Σ�����NB���������г��������ݴ����Բ��㡢���Ȳ���֡�ģ����·�������������⣬��ҵ����ǰ�������ļ��ݰ����빫������������ڹ�ͨ���ƣ��Խ������ʱ���벻���Ϸ��ա�ͬʱ��Ӧ����MDR Annex II/III��AIA Annex IV��ISO 13485�����IEC���ļ����ļ��Ϲ����ȷ���ĵ��ṹһ�¡�����������������ȷ���Ӹ��������NB���Ч�ʡ�
���⣬AIA������������Ҫ��ǿ�����ݼ��������ŷ��Ŀ����Ⱥ��һ���ԣ��й���ҵӦͨ��ŷ�˱��غ��������������ݼ�����ȷ����ģ�����ñ߽�������ģ�Ϳ��Ŷȡ�
����ڳ���ѧϰ��AI��Ӧͨ��PDCP����ģ���·�Χ�����ⴥ��ʵ�����ĴӶ����½���CE��֤�ķ��գ��������к�������Ư�Ƽ�⡢ƫ�м����ع����ƣ��γ������ıջ����չ�����ϵ��
��������������MDR/IVDR��AIA��ȫ����أ�ŷ�˼�ܷ�Χ�ѴӴ�ͳ��Ʒ��ȫ��չ���㷨����������������ϵͳ���ȡ��й���ҵԽ����ɷ���ӳ�䡢��ϵ������NBǰ�ù�ͨ����Խ����δ����ŷ��MDAI�г�������ռ��������λ��
����
����MDR/IVDR��AIA��ȫ����أ�ŷ���ѽ�MDAI����һ����߲�Ʒ��ȫ���㷨�������¼�ܽΡ���ҵ�ڽ���ŷ���г�ʱ������ͬʱ����MDR/IVDR��ȫ�ԡ��������ٴ�֤�ݵ�Ҫ���Լ�AIA���������������ȡ�����ල�����簲ȫ��ϵͳ������
�����й���ҵ���ԣ�������ż���Ҳ�ǻ�������ս���ڣ�ŷ�˼����ϵҪ����ҵ�ڲ�Ʒ����֮�⣬����߱���������������ܹ���ģ����֤��ϵ�����չ�����ϵ�����Ȼ��ƣ����������ĵ�������������CE��AIA˫����飬��ҵ������ISO 13485֮����������������ģ����֤��ƫ�п��ƺ����к���Ȼ��ƣ��������ǿ����������½ε�ȫ����AI�����Ϲ���ϵ�����ͬʱ��AIA��8��������һ�廯�����Ŀռ䣬ʹ��ҵ����ͨ��ͳһ������������ϵʵ�ָ��ߵĺϹ�Ч�ʡ�
����δ����MDAI�ľ������ƽ��������Լ������ܣ���������ҵ�ڷ��տ��ơ����ȡ������Լ������������������������Խ����� MDR/IVDR��AIA����ϵ�����ϣ�Խ����ŷ���г��л��ȷ���ԡ���ȫ���뾺���ԡ��Ϲ治��ֻ�����ż��������ڳ�Ϊȫ��ҽ��AI��ҵ�ĵײ㾺�����볤�ڳɹ��Ĺؼ�������
ע �ͣ�


